Bệnh thalassemia, còn được gọi là bệnh tan máu, được di truyền từ cha mẹ sang con cái. Bệnh gây hậu quả nặng nề về chủng tộc cũng như tính mạng của người bệnh trong cộng đồng. Tuy nhiên, bệnh có thể được ngăn ngừa bằng cách phát hiện sớm và can thiệp kịp thời. Bài viết dưới đây sẽ cung cấp cho bạn đọc những thông tin cơ bản nhất về căn bệnh này.
1. Thalassemia là gì?
Bệnh thalassemia (Thal) là một bệnh thiếu máu tán huyết di truyền hoặc thiếu máu tán huyết bẩm sinh. Triệu chứng chính của bệnh là thiếu máu.
Bệnh được chia thành hai loại chính:
– α(alpha) Thalassemia: Thiếu chuỗi α (alpha)
– β (beta) Thalassemia: Thiếu chuỗi β(beta)
Trong hai hình thức này, có 3 mức độ bệnh:
-Nặng;
– Trung gian;
-Ánh sáng.
2. Dịch tễ học của bệnh thalassemia
Bệnh thalassemia là một trong những bất thường di truyền phổ biến nhất trên thế giới. Hiện nay, 7% số người trên toàn cầu mang gen bệnh tan máu; 1,1% các cặp vợ chồng có nguy cơ sinh con mắc bệnh hoặc mang gen bệnh.
Bệnh phân bố trên toàn thế giới, với tỷ lệ lưu hành cao ở khu vực Địa Trung Hải, Trung Đông và khu vực châu Á – Thái Bình Dương. Việt Nam là một trong những quốc gia có tỷ lệ mắc bệnh và bệnh di truyền cao trong khu vực. Tại Việt Nam, tỷ lệ gen bệnh ở người Kinh khoảng 2 – 4%, ở đồng bào dân tộc thiểu số sống ở miền núi, tỷ lệ này khá cao, như: 22% đối với dân tộc Mường, và trên 40% đối với dân tộc Mường. E-de, Tày, Thái, Stieng…
3. Biểu hiện của bệnh thalassemia
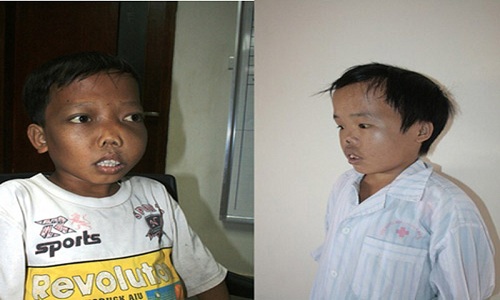
3.1 Mức độ nghiêm trọng
Biểu hiện của thiếu máu nghiêm trọng, có thể xảy ra ngay sau khi sinh, thường rõ ràng nhất khi bé được 4 – 6 tháng tuổi và trở nên tồi tệ hơn.
Các triệu chứng thường gặp bao gồm:
– Trẻ nhợt nhạt;
– Da vàng và màng cứng của mắt;
– Thường chậm phát triển thể chất;
– Có thể bị sốt, tiêu chảy hoặc các rối loạn tiêu hóa khác.
Với việc truyền máu đầy đủ, trẻ em có thể tiếp tục phát triển bình thường cho đến khoảng 10 tuổi. Sau 10 tuổi, trẻ có các biến chứng do tăng hồng cầu và tích tụ quá nhiều sắt trong cơ thể như:
– Biến dạng xương: hộp sọ lớn, bướu trán, bướu đỉnh, xương gò má cao, mũi phẳng, răng cửa hàm trên nhô ra, loãng xương khiến trẻ rất dễ bị gãy xương;
– Da xỉn màu, màng cứng màu vàng;
– Sỏi mật;
– Dậy thì muộn: bé gái đến 15 tuổi chưa có kinh nguyệt…;
– Chậm phát triển thể chất. Sau 20 tuổi, bệnh nhân thường có nhiều biến chứng suy tim, rối loạn nhịp tim, tiểu đường, xơ gan…
3.2. Mức trung bình
Biểu hiện thiếu máu xuất hiện muộn hơn mức độ nghiêm trọng, trẻ khoảng 4 – 6 tuổi cần truyền máu. Thiếu máu vừa hoặc nhẹ (nồng độ hemoglobin 6g / dl đến 10g / dl). Tuy nhiên, nếu không được điều trị đầy đủ và kịp thời, bệnh nhân cũng sẽ có các biến chứng như lá lách to, gan to, sỏi mật, sạm da. Đến tuổi trung niên sẽ có các triệu chứng tiểu đường, suy tim, xơ gan. Nếu bệnh nhân được truyền máu đầy đủ và thải sắt, người đó có thể phát triển bình thường và không có biến chứng.
3.3. Mức độ nhẹ (hay còn gọi là mang gen)
Người mang mầm bệnh thường không có bất kỳ biểu hiện lâm sàng đặc biệt nào. Chỉ trong những giai đoạn cơ thể có nhu cầu máu tăng cao như phụ nữ khi mang thai, kinh nguyệt nặng, v.v., chúng ta mới thấy dấu hiệu mệt mỏi, da xanh, nếu chúng ta làm xét nghiệm, chúng ta sẽ thấy lượng huyết sắc tố. giảm.
4. Bệnh thalassemia được điều trị như thế nào?
Hiện nay, các phương pháp chính để điều trị bệnh Thalassemia là:
4.1. Điều trị thiếu máu: Truyền hồng cầu khi bệnh nhân bị thiếu máu, với nồng độ hemoglobin là 7g/dl sau 2 lần khám mà không có nguyên nhân nào khác, hoặc >7g/dl bị biến dạng xương. Để ngăn ngừa và kiểm soát các tác dụng không mong muốn có thể xảy ra do truyền máu, bệnh nhân cần được theo dõi chặt chẽ trong bệnh viện.
4.2. Điều trị quá tải sắt: Thải sắt bằng đường tiêm hoặc uống. Cần bắt đầu thải sắt khi ferritine huyết thanh > 800 ng/ml, thường là sau khi truyền khoảng 20 đơn vị máu lắng. Liệu pháp thải sắt suốt đời.
4.3. Cắt lách: Chỉ khi truyền máu kém hiệu quả hoặc lá lách quá to, gây đau đớn và ảnh hưởng đến sinh hoạt của người bệnh.
4.4. Ghép tế bào gốc tạo máu (ghép tủy): Áp dụng cho bệnh nhân bệnh nặng, là phương pháp tiên tiến nhất hiện nay có thể chữa khỏi bệnh. Tại Việt Nam, một số bệnh viện hàng đầu trong ngành có thể thực hiện phương pháp điều trị này, tuy nhiên hạn chế của phương pháp này là khó tìm được người hiến tế bào gốc phù hợp.
5. Tự chẩn đoán sớm bệnh Thalassemia?
– Bệnh nhân thalassemia thừa hưởng 2 gen bệnh (trong Beta thalassemia) hoặc 3-4 gen bệnh (trong Alpha thalassemia) từ cha mẹ. Tan máu xảy ra liên tục trong suốt cuộc đời, gây thiếu máu mãn tính và tích tụ sắt trong cơ thể. Bệnh nhân thường có biểu hiện thiếu máu tán huyết, vàng da, lách to, thường cần truyền máu định kỳ và thải sắt suốt đời.
– Người mang mầm bệnh thalassemia thừa hưởng 1 gen bệnh (trong Beta thalassemia) hoặc 1-2 gen bệnh (trong Alpha thalassemia) từ cha mẹ. Những người này không có biểu hiện lâm sàng, họ có sự phát triển trí tuệ và thể chất bình thường. Nhiều nhà khoa học, phi công, kỹ sư và bác sĩ là những người mang gen Thalassemia. Số người mang gen bệnh trên thế giới rất cao (hơn 400 triệu người), ở nước ta ước tính con số này là 10 triệu người. Những người mang gen bệnh trong suốt cuộc đời sẽ không phát triển bệnh. Phòng ngừa bệnh thalassemia nên được thực hiện tại thời điểm kết hôn và sinh con.
Nếu hai người mang gen kết hôn, có 25% khả năng đứa trẻ sẽ bị bệnh thalassemia nặng khi có con, 50% khả năng đứa trẻ sẽ mắc bệnh nhẹ hoặc là người mang mầm bệnh; 25% khả năng em bé bình thường.
6. Sàng lọc người mang gen thalassemia
6.1 Đối tượng
– Tất cả người nhà của bệnh nhân bị bệnh Thalassemia.
– Người thực hiện phân tích máu toàn bộ với thiếu máu vi hồng cầu hypochromic (MCV < 85 fL và / hoặc MCH < 28 pg).
Những người đang có kế hoạch kết hôn và sinh con cũng nên được sàng lọc bệnh Thalassemia.
6. 2 Xét nghiệm
– Phân tích tế bào máu ngoại vi: Hemoglobin giảm hoặc bình thường, giảm hồng cầu nhỏ (MCV < 85fL và/hoặc MCH < 28pg), với hình thái và kích thước đa dạng… Với kết quả Với điều này, bác sĩ có thể chẩn đoán sơ bộ bạn là có hoặc mang gen Thalassemia.
– Định lượng sắt huyết thanh, ferritin huyết thanh và tranferin huyết thanh để loại trừ thiếu máu do thiếu sắt.
– Điện di huyết sắc tố: là phương pháp chẩn đoán bệnh thalassemia.
– Xét nghiệm di truyền có thể xác định chính xác các đặc điểm của tổn thương gen tổng hợp chuỗi globin trong bệnh thalassemia.
7. Làm thế nào để ngăn ngừa bệnh thalassemia?
– Tư vấn tiền hôn nhân: Các cặp vợ chồng cần được khám và xét nghiệm Thalassemia trước khi kết hôn.
– Tất cả thân nhân của bệnh nhân mắc bệnh Thalassemia cần được sàng lọc bệnh thalassemia.
– Nếu cả hai người mang cùng gen Thalassemia đã kết hôn, nên tìm tư vấn trước khi lên kế hoạch mang thai.
– Nếu vợ chồng mang cùng gen Thalassemia đang mang thai thì cần được chẩn đoán trước khi sinh ở tuần thai 12-18 tại các cơ sở y tế chuyên khoa.
– Nên được tư vấn bởi các bác sĩ chuyên khoa huyết học, nhi khoa và di truyền học về bệnh thalassemia.
Khi phát hiện bệnh nhân Thalassemia hoặc người mang mầm bệnh, tất cả các thành viên trong gia đình nên được sàng lọc bệnh thalassemia.